Ataxin 1 antibody
Cat. No. GTX87190
Cat. No. GTX87190
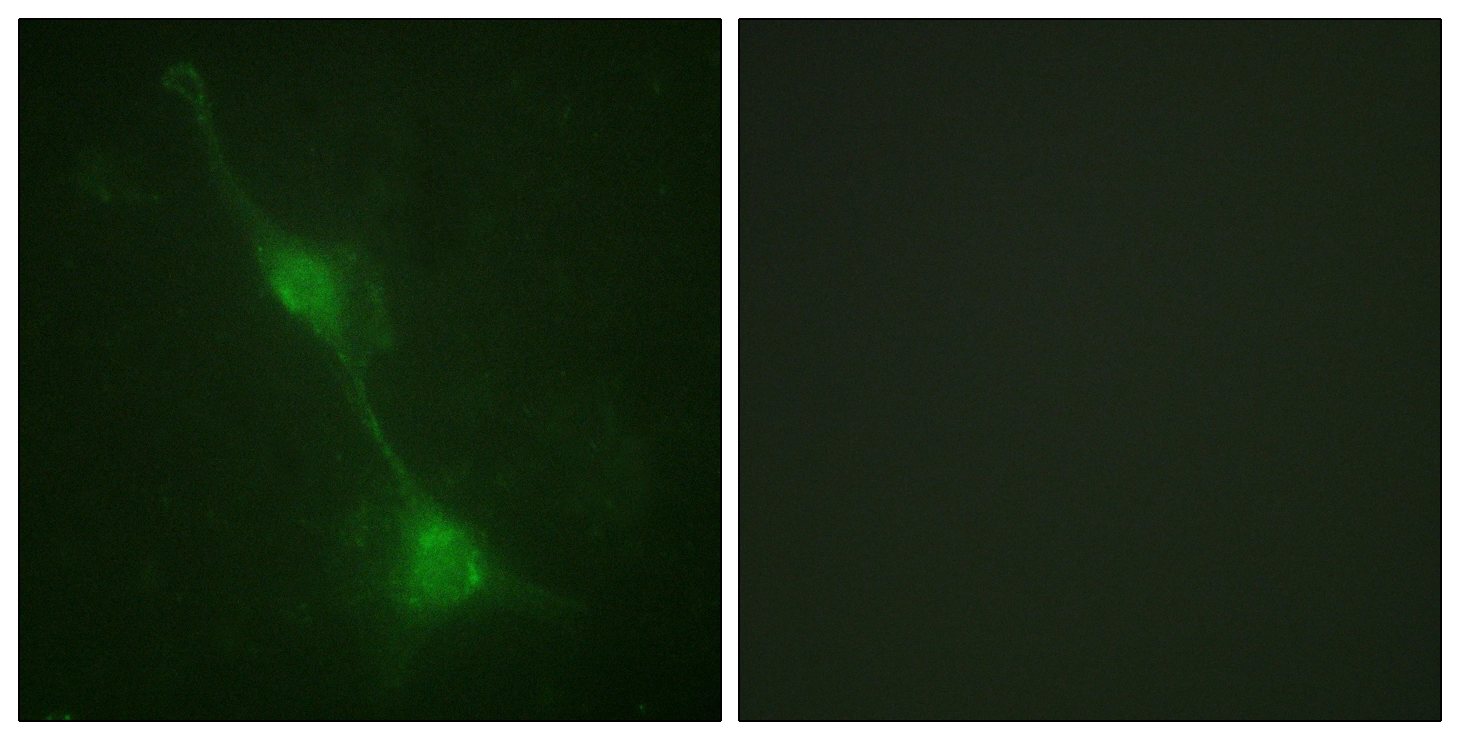
GTX87190 ICC/IF Image
ICC/IF analysis of NIH3T3 cells using GTX87190 Ataxin 1 antibody. The picture on the right is blocked with the synthesized peptide.
1 / 2
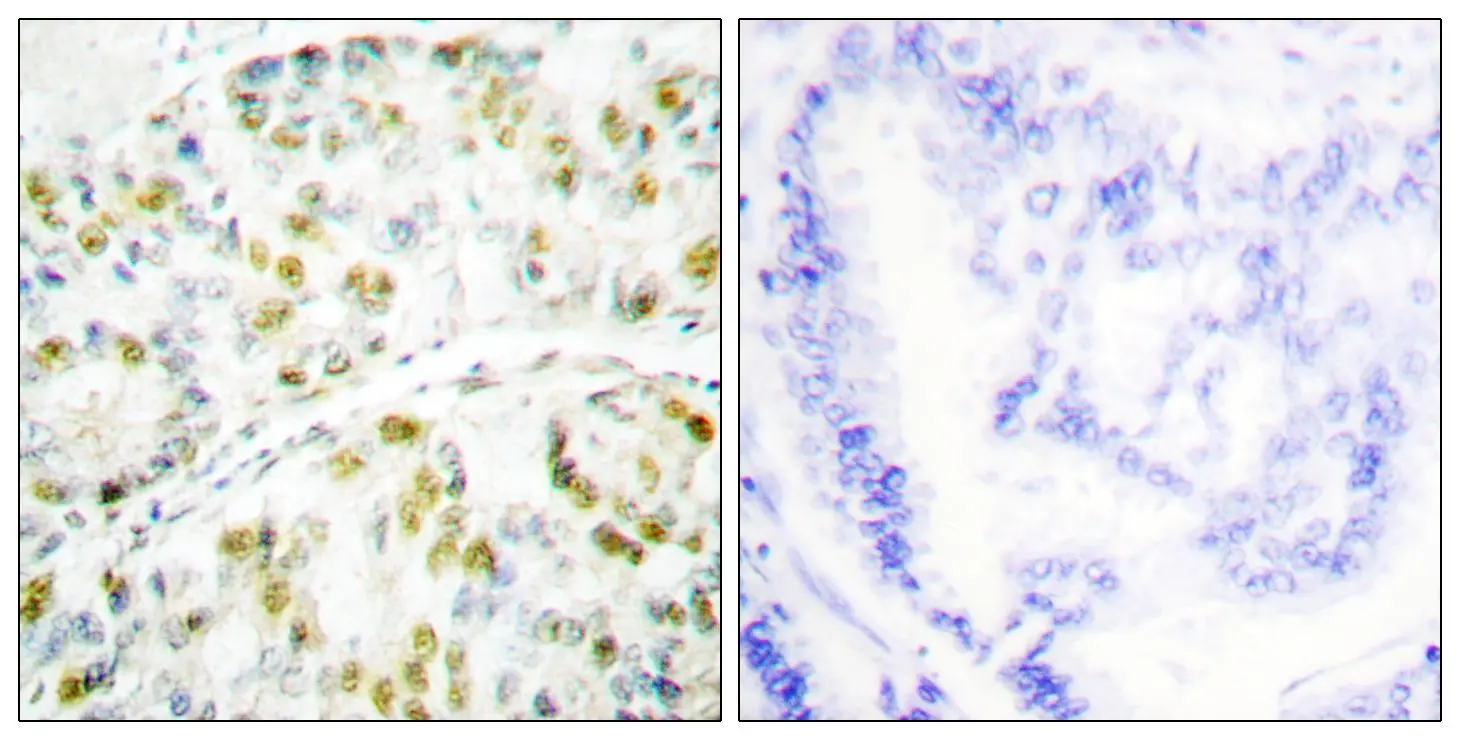
GTX87190 IHC-P Image
IHC-P analysis of human lung carcinoma tissue using GTX87190 Ataxin 1 antibody. The picture on the right is blocked with the synthesized peptide.
2 / 2
-
HostRabbit
-
ClonalityPolyclonal
-
IsotypeIgG
-
ApplicationsICC/IF IHC-P
-
ReactivityHuman, Mouse