CFTR antibody

WB analysis of rat lung membrane lysate using GTX54774 CFTR antibody preincubated with or without immunogen peptide.
Dilution : 1:200
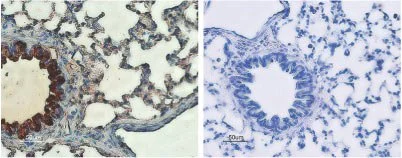
IHC-P analysis of rat lung tissue using GTX54774 CFTR antibody. Strong staining of bronchial epithelial cells (red) and lighter staining of alveolar cells (red-brown) is apparent. There is also positive staining of macrophages while smooth muscle and endothelium are negative. Counterstain of cell nuclei appears blue. A negative control is shown in the right panel.
-
HostRabbit
-
ClonalityPolyclonal
-
IsotypeIgG
-
ApplicationsWB ICC/IF IHC-P IP
-
ReactivityHuman, Mouse, Rat