Human GALE protein, His tag
Cat. No. GTX67401-pro
Cat. No. GTX67401-pro
GTX67401-pro Image
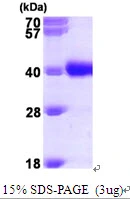
3μg Human GALE protein (GTX67401-pro) by SDS-PAGE under reducing condition and visualized by coomassie blue stain.
1 / 1
-
SpeciesHuman