Human NEU1 protein, His tag
Cat. No. GTX00129-pro
Cat. No. GTX00129-pro
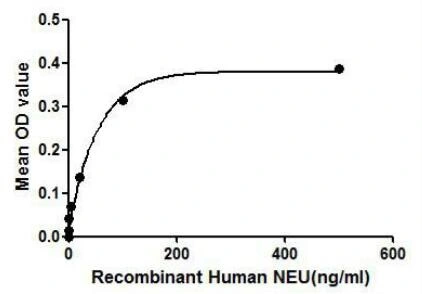
GTX00129-pro Functional Assay Image
Functional ELISA analysis of GTX00129-pro Human NEU1 protein which can bind immobilized CTSA protein.
1 / 3
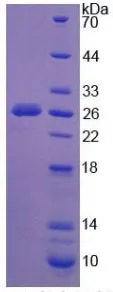
GTX00129-pro Image
SDS-PAGE analysis of GTX00129-pro Human NEU1 protein.
2 / 3
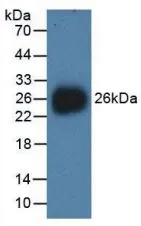
GTX00129-pro Image
WB analysis of GTX00129-pro Human NEU1 protein.
3 / 3
-
ApplicationsFunctional Assay
-
SpeciesHuman